What is Johanson-Blizzard Syndrome?
Introduction
History
Causes and symptoms
Genetics
Epidemiology
Case reports
Prognosis and treatment
References
Further reading
Johanson–Blizzard syndrome (JBS) is a rare autosomal recessive condition caused by homozygous and compound heterozygous mutations in the UBR1 gene. Until 2005, the genetic abnormality that caused the syndrome was unknown.
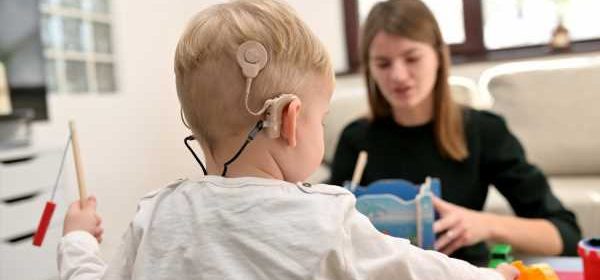
Multisystem involvement and facial dysmorphic characteristics are hallmarks of this syndrome. Bilateral severe to profound sensorineural hearing loss is one of the most common symptoms in JBS patients. Microcephaly, hypoplasia of the alae nasi, absence of permanent teeth, and scalp abnormalities are some of the dysmorphic symptoms of this condition. The intensity, indications, and symptoms of JBS differ from person to person. Many symptoms can be seen as early as birth or in infancy.
 Bilateral severe to profound sensorineural hearing loss is one of the most common symptoms in JBS patients. Image Credit: mady70/Shutterstock
Bilateral severe to profound sensorineural hearing loss is one of the most common symptoms in JBS patients. Image Credit: mady70/Shutterstock
History
In 1971, Johanson and Blizzard described three unrelated patients with malabsorption due to exocrine pancreatic insufficiency (EPI), as well as a variety of other clinical features such as nasal alae hypoplasia or aplasia, scalp defects, imperforate anus, deafness, hypothyroidism, dental defects, genitourinary malformations, and learning disabilities. Fox et al. in 1976 coined the term Johanson–Blizzard syndrome (JBS) to describe this condition five years later.
Causes and symptoms
Mutations in the UBR1 gene cause Johanson-Blizzard syndrome. This gene instructs the body to make a protein that is necessary for the pancreas to function properly. Acinar cells, which are found in the pancreas, generate this protein. Acinar cells are crucial because they assist the pancreas to create digestive enzymes that allow it to break down food and use the nutrients for growth.
The intensity, indications, and symptoms of JBS differ from person to person. Many symptoms can be seen as early as birth or in infancy. A majority of patients (80 to 99%) experience symptoms including underdeveloped nasal alae, abnormal hair pattern, exocrine pancreatic insufficiency (EPI), short stature, and alopecia. Around 30-79% of patients with JBS have abnormal vagina morphology, anemia, anal atresia, delayed eruption of teeth and delayed skeletal maturation. They may also develop hypoproteinemia, microdontia, oligodontia, sensorineural hearing impairment, and intellectual disability. Abnormal cardiac septum morphology, cholestasis, dextrocardia, dilated cardiomyopathy, hepatic failure, and hydronephrosis are observed in 5% to 29% of patients.
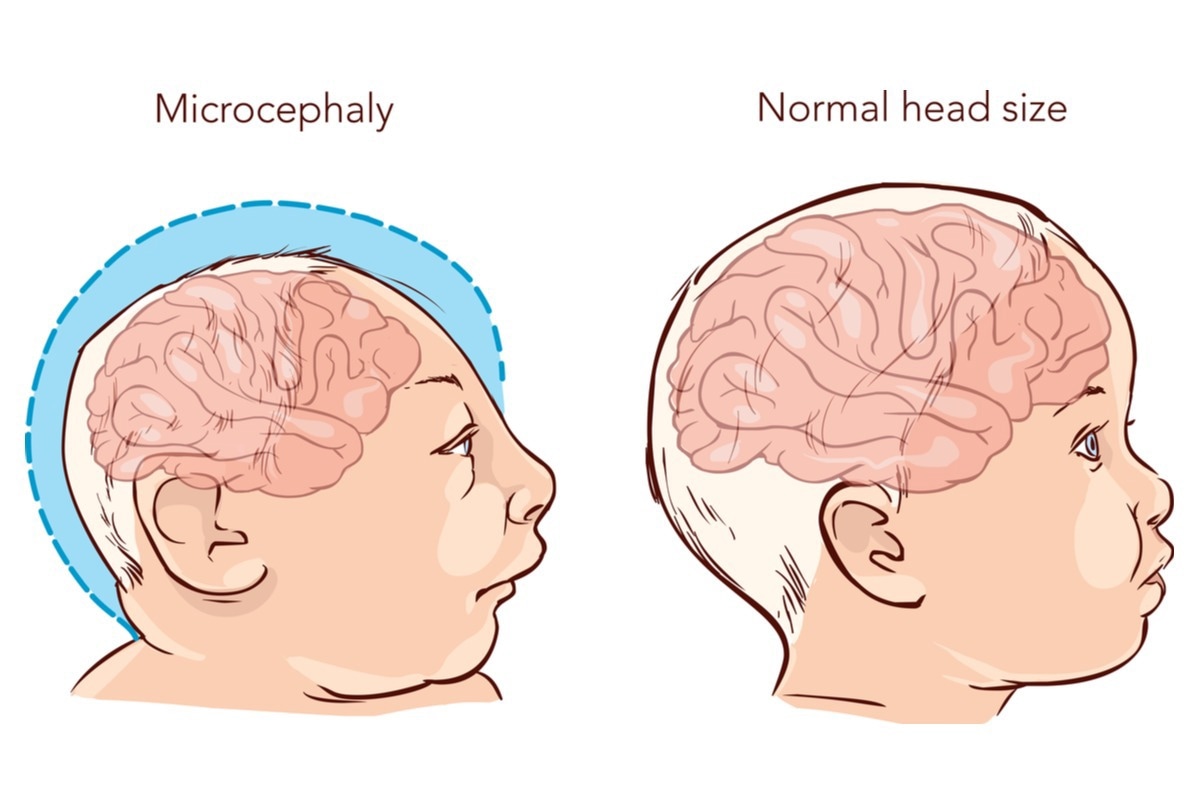 Microcephaly is another common symptom of JBS. Image Credit: Drp8/Shutterstock
Microcephaly is another common symptom of JBS. Image Credit: Drp8/Shutterstock
Genetics
The gene UBR1 is found on chromosome 15q15-21. UBR1 is one of at least four functionally overlapping E3 ubiquitin ligases in the N-end rule pathway, a conserved proteolytic mechanism with destabilizing N-terminal residues 20 as substrates. When persons with Johanson-Blizzard syndrome have a malfunctioning UBR1 gene, the pancreas' acinar cells are damaged, and the pancreas is unable to break down lipids and other nutrients. Many of the signs of JBS, such as sluggish growth, are caused as a result of this. In people with JBS, the ubiquitin ligase gene (UBR1) has nonsense, frameshift, and splice-site mutations. This syndrome follows the autosomal recessive pattern of inheritance.
Epidemiology
JBS is predicted to affect 1 in every 250,000 people. There is no gender difference in JBS cases reported to date. Consanguinity between parents is a common occurrence.
Case reports
The clinical characteristics of four patients with JBS and significant oblique facial clefts (OFCs) were documented in a paper published by Riverra et al., in 2016. The presence of homozygous or compound heterozygous mutations in the UBR1 gene validated the diagnosis of JBS in all of them. In addition, we look at three earlier occurrences of JBS involving OFCs. We estimate that the frequency of OFCs in JBS is between 5% and 10% based on about 100 patients affected by JBS who have been published in the literature. The severity of extensive OFCs may be at the end of the spectrum of facial malformations seen in JBS, according to this study. Within this cohort, no obvious genotype-phenotype correlation could be found. As a result, UBR1 should be included in the list of OFC contributing genes, though the particular mechanism is unknown.
Sood et al., in 2018 presented a case of a child with pancreatic insufficiency and Johanson-Blizzard syndrome-like facial abnormalities. Fetal ultrasound revealed ambiguous genitalia in a 21-year-old G1P1 lady. A phallic structure with urethral meatus, non-palpable gonads, and two orifices in close proximity in the perineum, the anterior being a common urogenital channel and the posterior being the rectum, was discovered at birth. A voiding cystourethrogram/genitogram revealed bilateral high-grade vesicoureteral reflux and a 1.5-cm-long common urogenital sinus that split into three channels: the native urethra, an auxiliary urethra pointing anteriorly towards the clitoris, and a septate vagina with uterus didelphys. The clinical presentation suggested JBS, which was validated by UBR1 molecular testing (46, XX). She had feminizing genitoplasty and posterior sagittal anorectoplasty at the age of 16 months.
 Related: What is Jackson-Weiss Syndrome?
Related: What is Jackson-Weiss Syndrome?
Prognosis and treatment
The symptoms of Johanson-Blizzard syndrome (JBS) vary in severity. While some people acquire life-threatening complications throughout childhood, others have a milder condition. Although intellectual disability does exist, intelligence can also be normal in some situations. Hearing loss or deafness can occur at any age or develop later in life. Malabsorption and exocrine pancreatic insufficiency may cause growth deficiency in JBS patients.
If left untreated, pancreatic dysfunction and malabsorption can lead to life-threatening complications such as infections and malnutrition. A better prognosis can be achieved by treating pancreatic insufficiency and hypothyroidism separately. Life expectancy is highly dependent on pancreatic insufficiency and malnutrition, which can lead to death in infancy or early childhood, but survival into adulthood is not uncommon in patients who are treated early with effective pancreatic enzymes and vitamin replacement.
Johanson-Blizzard syndrome is treated by focusing on the specific symptoms that each person experiences. Pancreatic insufficiency may necessitate the use of pancreatic enzyme supplements to ensure normal fat and other nutrient absorption, as well as vitamin supplements to prevent or correct vitamin deficits caused by malabsorption due to pancreatic insufficiency.
The removal of the pancreas (pancreatectomy) and islet auto-transplantation (TPIAT) is only done as a last resort for patients with severe pancreatitis symptoms. Other anomalies connected with the illness, such as craniofacial, genitourinary, cardiac, and/or other deformities, may be corrected with surgery. Bonding chemicals, dentures, and/or other procedures may be used to address dental problems. Hearing aids can help those with hearing loss. To guarantee that children with JBS attain their full potential, early intervention is critical. Special remedial education, special social assistance, and other medical, social, and/or vocational services may be beneficial to affected children.
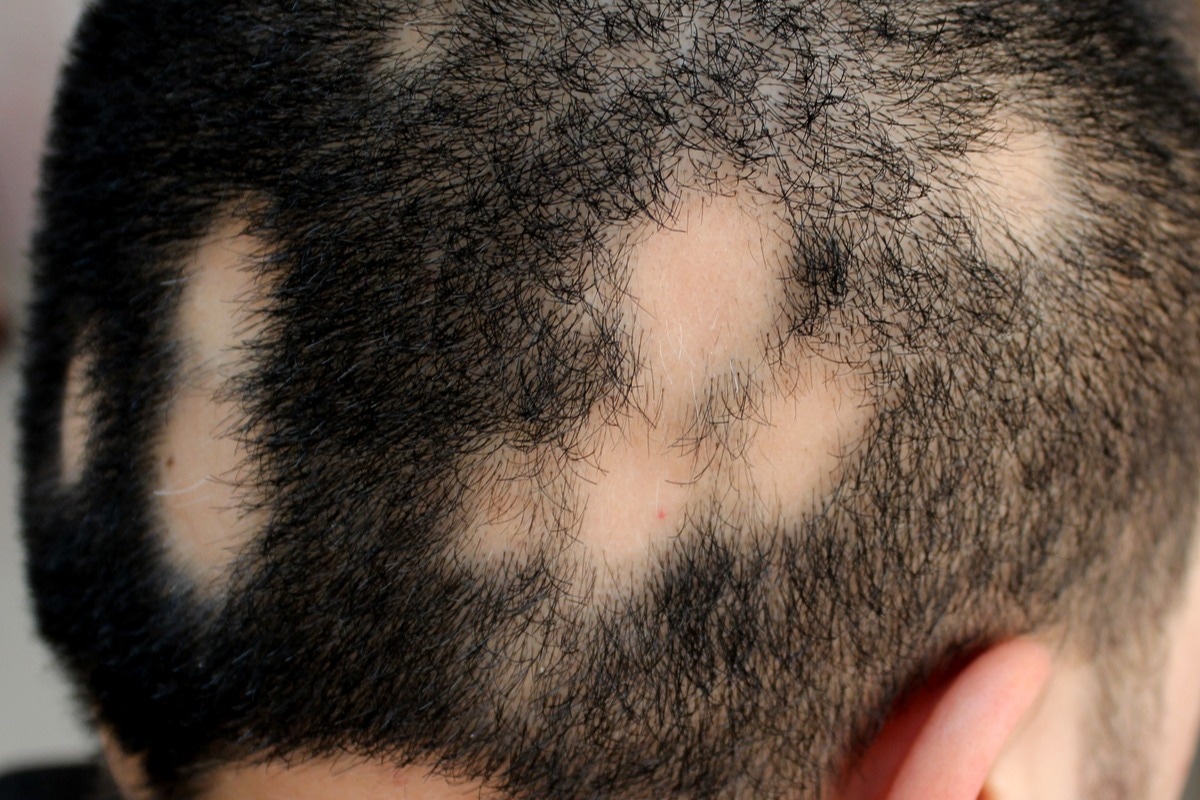 Alopecia is a symptom in over 80% of JBS cases. Image Credit: Dermatology11/Shutterstock
Alopecia is a symptom in over 80% of JBS cases. Image Credit: Dermatology11/Shutterstock
References
- Johanson-Blizzard syndrome. [Online] NIH0GARD. Available at: https://rarediseases.info.nih.gov/diseases/80/johanson-blizzard-syndrome
- JOHANSON-BLIZZARD SYNDROME; JBS. [Online] OMIM. Available at: https://www.omim.org/entry/243800
- Almashraki, N., Abdulnabee, M. Z., Sukalo, M., Alrajoudi, A., Sharafadeen, I., & Zenker, M. (2011). Johanson-Blizzard syndrome. World journal of gastroenterology, 17(37), 4247–4250. https://doi.org/10.3748/wjg.v17.i37.4247
- Ellery, K. M., & Erdman, S. H. (2014). Johanson-Blizzard syndrome: expanding the phenotype of exocrine pancreatic insufficiency. JOP: Journal of the pancreas, 15(4), 388–390. https://doi.org/10.6092/1590-8577/2409
- Corona-Rivera, J. R., Zapata-Aldana, E., Bobadilla-Morales, L., Corona-Rivera, A., Peña-Padilla, C., Solis-Hernández, E., Guzmán, C., Richmond, E., Zahl, C., Zenker, M., & Sukalo, M. (2016). Oblique facial clefts in Johanson-Blizzard syndrome. American journal of medical genetics. Part A, 170(6), 1495–1501. https://doi.org/10.1002/ajmg.a.37630
- Sood, A., Jamzadeh, A., Chowdhury, M., & Lakshmanan, Y. (2018). Johanson-Blizzard syndrome with associated urogenital anomalies. BMJ case reports, 2018, bcr2018225336. https://doi.org/10.1136/bcr-2018-225336
- Alwadee, R. M., Alyousef, M. Y., Yousef, E. M., & Yousef, M. F. (2021). Performance of Children with Johanson-Blizzard Syndrome After Cochlear Implantation. Cureus, 13(11), e19264. https://doi.org/10.7759/cureus.19264
Further Reading
- All Rare Disease Content
- What is a Rare Disease?
- Importance of Research into Rare Disease
- Teaching old drugs new tricks – drug repurposing for rare diseases
- What is Adenomyosis?
Last Updated: Jul 7, 2022

Written by
Aimee Molineux
Aimee graduated from Oxford University with an undergraduate degree in Japanese and Korean Studies, with an exchange year at Kobe University in Hyogo, Japan. Throughout her studies, Aimee took part in various internships, gaining an interest in marketing and editorial work along the way.In her personal time, Aimee can be found either attempting to cook, learning how to code, doing pilates, as well as regularly updating her pet hamster’s Instagram account.
Source: Read Full Article