Buy estrace canadian pharmacy without prescription
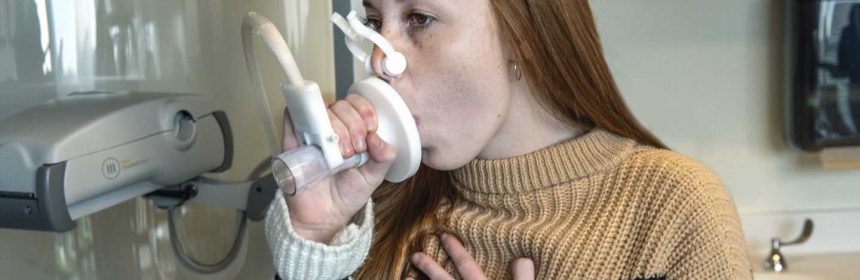

Anna Thompson looks like a healthy 15-year-old, but the high school student has been living with cystic fibrosis since birth. Her daily regimen includes medications, pancreatic enzymes, nebulizer treatments and chest physical therapy three times a day with a vest that pulses air in and vibrates her chest.
“You wouldn’t know anything is wrong with her if you met her,” said her father, Eddie Thompson. “She’s been really good and responsible about keeping her treatments up. She’s been a relatively healthy cystic fibrosis patient.”
Anna tested positive for cystic fibrosis when she was six days old. “From that point on we were at Children’s Hospital of Richmond at VCU probably once a week for a while, then once every couple of weeks, and then once a month for scheduled checkups,” Thompson said. “When she was two, we went once every three months.”
Along with her daily treatments, Anna also has to monitor her weight and follow a high-calorie diet. “She struggles to keep her weight up,” her dad said.
Even though she’s been diligent about her daily routine, Anna and her parents still worry about the future.
Cystic fibrosis is a progressive disease caused by a defective protein that results from two mutations in the cystic fibrosis regulator gene. The disease causes the formation of thick mucus that builds up in the lungs, digestive tract and other parts of the body. It leads to severe respiratory and digestive problems as well as other complications such as infections and diabetes.
The cystic fibrosis community received news in October that the Food and Drug Administration approved a new game-changing drug, Trikafta. The news was particularly exciting for patients like Anna who previously had no therapeutic drug options for their specific cystic fibrosis mutation. Previously, only patients with a rare mutation qualified for drug therapies but this drug dramatically improves quality of life and works for the majority of cystic fibrosis patients—the 90 percent who have this particular, more common mutation. Anna has been on the drug for three weeks and is doing well.
Trikafta is approved for patients 12 and older with cystic fibrosis who have at least one F508del gene mutation, which is estimated to represent about 27,000 people in the U.S., according to the FDA.
CHoR was one of only 44 centers in four countries testing the drug with 510 cystic fibrosis patients 12 and older. It was an “honor to be accepted into this trial. It was an elite group that was participating,” said Joel Schmidt, M.D., pediatric program director for the Cystic Fibrosis Center at CHoR and a principal investigator for one of the Trikafta clinical trials.

The clinical trials leading to the FDA’s approval in October were “built on a lot of research,” Schmidt said. “Ninety percent of [cystic fibrosis] patients have one copy of the F508del gene and that’s what this drug targets. It’s remarkable.”
Some patients in the trials had increased lung function, on average from 10% to 13%, something that never has been achieved before. They also saw improvements in sweat chloride (amount of salt in sweat), body mass index and a decrease in respiratory symptoms.
“One patient that started the drug six weeks ago in a compassionate use program before the FDA approval was getting ready for a transplant,” Schmidt said. “After six weeks, she was no longer on the transplant list. Patients also reported better quality of life on the medicine.”
Meg Lessard, senior clinical research coordinator in the Pediatric Research Office at CHoR, and Ryan Hayden, clinical research and lead coordinator in the Trikafta clinical trial, have been conducting tests and assessing patients. “We make sure they stay engaged with the clinical and research teams,” Lessard said. “We make sure the research protocol is being followed for patient safety and to ensure data quality.”
They have seen an increase in patients’ pulmonary function test after taking the drug. Patients also have reported that they “just feel better,” Lessard said.
“It’s not as if this drug is a one-time cure. The drug doesn’t ‘fix’ the inherited mutations, but rather dramatically improves the functioning of the mutated … proteins,” Hayden added. “Patients are feeling better and getting sick less.”
Clinical trials for younger children ages six to 11 are still ongoing, Lessard said. “The hope is to extend trials to even younger children in order to improve and preserve lung function early on in life.”
Trikafta will reach more people with cystic fibrosis—90 percent—than any other drug before and is expected to add decades to a child’s life.
Source: Read Full Article