What impact has SARS-CoV-2 variant B.1.1.519 had on Mexico City?

Coronavirus disease 2019 (COVID-19) has become one of the most concerning diseases globally since its initial outbreak. It is of great importance that monitoring new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants is ongoing because mutations could change viral biological properties. Viral genome alterations could alter neutralization efficiency resulting from naturally acquired immunity, and there may be differences in the clinical presentation of the disease.
 Study: The evolutionary landscape of SARS-CoV-2 variant B.1.1.519 and its clinical impact in Mexico City. Image Credit: Eve Orea/ Shutterstock
Study: The evolutionary landscape of SARS-CoV-2 variant B.1.1.519 and its clinical impact in Mexico City. Image Credit: Eve Orea/ Shutterstock
The SARS-CoV-2 B.I.I.519 variant was responsible for 90% of cases detected in February 2021 in Mexico City. This variant is characterized by three amino acid changes in the spike protein (P681H, T478K, and T732A).
The current study reports the effective reproduction number of B.1.1.519 and presents its geographical origin based on phylogenetic analysis. Also investigated was this variants evolution through haplotype analysis, and the most recent haplotypes were identified. Finally, the authors examined the clinical impact of patients infected with variant B.1.1.519 compared to individuals infected with non-B.1.1.519 SARS-CoV-2.
A preprint version of the study is available on the medRxiv* server while the article undergoes peer review.
Variant identification in Mexico City
The first patient infected with the B.1.1.519 variant in Mexico City was identified on November 3, 2020, only the second case recorded worldwide. Within Mexico City, the variant frequency increased from 16% to 90% in February 2021 but decreased to 51% in May 2021.
Of sequences generated from SARS-CoV-2 infections, variant B.1.1.519 represented 74.3% from November 2020 to May 2021 in Mexico City. The B.1.1.519 variant was detected in 31 countries, making up 55% of cases in Mexico. The B.1.1.519 variant is grouped in an independent clade derived from the clade 20B NextClade classification according to phylogenetic analysis. The B.1.1.519 variant is characterized by nine mutations, four of which are ORF1 substitutions and three spike substitutions.
Reproduction number
To determine the transmissibility of variant B.1.1.519, the authors studied the effective reproduction number (Rt), which was defined as the average number of secondary cases per primary case at a given time within the year. It was observed that there was a sharp increase in the Rt for the B.1.1.519 variant, up to a value of 2.9 in the second week of December 2020, which corresponded with the increased detection. However, the Rt value began to stabilize in the following months, between 0.5 and 1.
The B.1.1.519 variant was the second most frequently detected in Mexico City. The Rt value for variant B.1.1.519 fluctuated strongly in the months following December, which may have been influenced by the small number of cases associated with this variant. This strong fluctuation differs from other variants detected in that region as other variants stabilized or disappeared as the year progressed.
Genomic findings
The maximum phylogeny was calculated, which included all SARS-CoV-2 genomes of interest to examine the geographic origin of variant B.1.1.519 and its evolutionary relationship to the B.1.1.222 variant. There are three defined clusters shown in the phylogenetic tree. Two only correspond to the B.1.1.519 and B.1.1.222 variants, with clear separation and a mixed cluster, displaying undefined separation among lineages. This demonstrates that the evolution of this SARS-CoV-2 variant lineage remains unclear, although the mixed cluster formed by variant B.1.1.519 sequences is most closely related to variant B.1.1.222 sequences.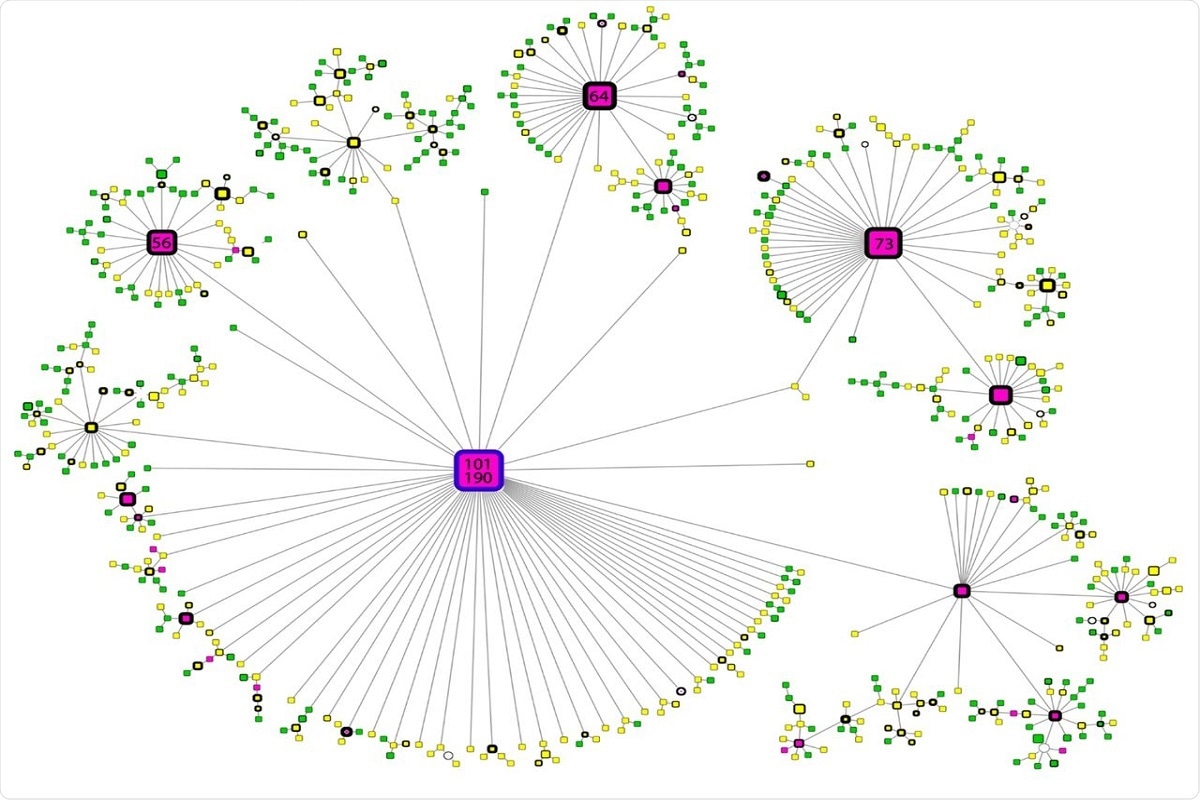 Figure 4. Haplotype network of B.1.1.519 sequences. Node colors represent the month of appearance (pink: November or December, yellow: January, February or March; green: April or May). Node size is proportional to the number of samples for that specific haplotype, and border width is proportional to the prevalence of the haplotype. Numbers correspond to the number of samples for that specific haplotype. The blue-bordered node indicates the haplotype with the most ancient appearance date for lineage B.1.1.519.
Figure 4. Haplotype network of B.1.1.519 sequences. Node colors represent the month of appearance (pink: November or December, yellow: January, February or March; green: April or May). Node size is proportional to the number of samples for that specific haplotype, and border width is proportional to the prevalence of the haplotype. Numbers correspond to the number of samples for that specific haplotype. The blue-bordered node indicates the haplotype with the most ancient appearance date for lineage B.1.1.519.
Clinical association
The authors studied the clinical impact of the B.1.1.519 variant by analyzing associations between the variant and the number of clinical traits. The only sequences considered for the analyses were those with complete sets of clinical data (N=600). Evaluation of the data revealed that patients infected with the B.1.1.519 variant displayed a significant increase in the likelihood of developing symptoms affecting the respiratory tract relative to non-B.1.1.519 variants.
Logistic regression models adjusted for viral cycle threshold, number of comorbidities, age, and sex revealed that B.1.1.519 variant was associated with a 1.786-fold increase in shortness of breath, a 1.489-fold increase in chest pain, and a 3.655-fold increase in cyanosis. Also, variant B.1.1.519 was associated with a higher fraction of patients developing serious illness or death.
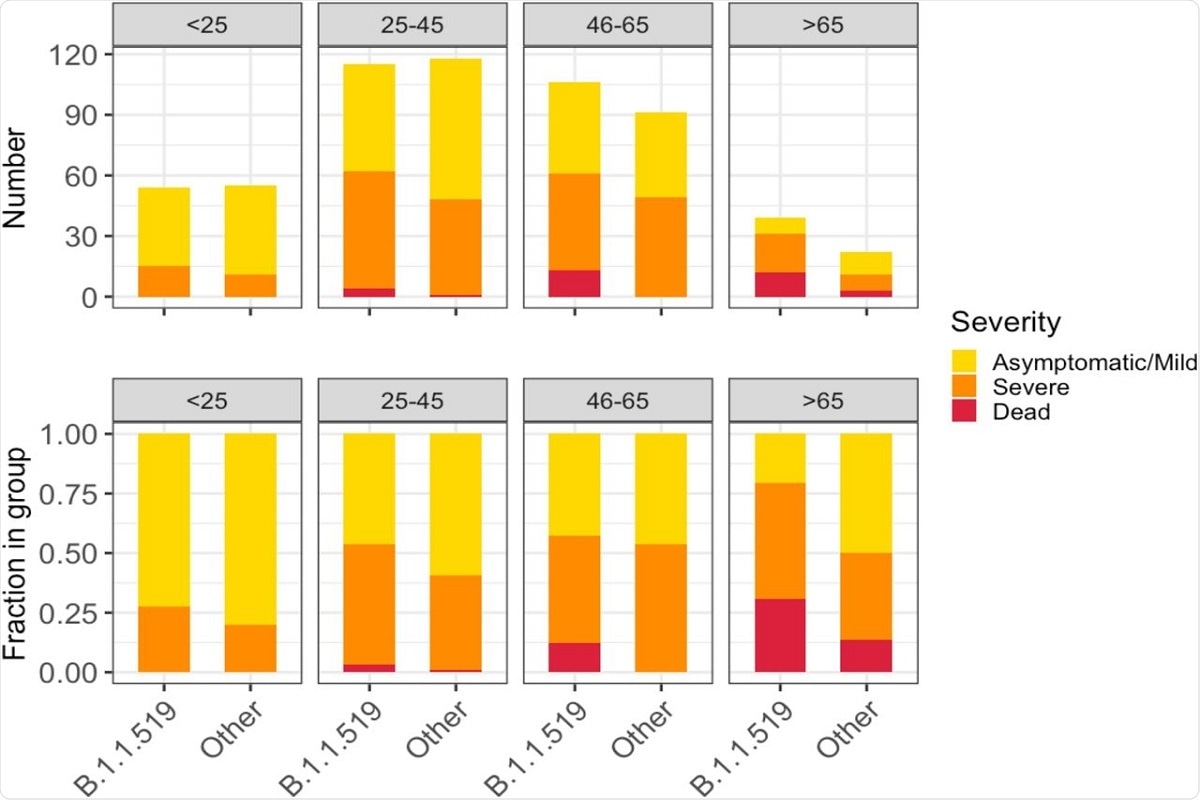 Figure 5. Severity of illness across patient age groups and by presence of B.1.1.519 or non-B.1.1.519 SARS-CoV-2 infections. The figure shows absolute counts (upper) and proportions of patients (lower).
Figure 5. Severity of illness across patient age groups and by presence of B.1.1.519 or non-B.1.1.519 SARS-CoV-2 infections. The figure shows absolute counts (upper) and proportions of patients (lower).
Implications
The authors observed that the B.1.1.519 SAR-CoV-2 variant was significantly associated with hospitalization, severe disease, and death. Patients infected with variant B.1.1.519 also appeared to show a higher prevalence of symptoms such as shortness of breath, chest pains, and cyanosis.
The more severe outcomes associated with variant B.1.1.519 are similar to those shown with the delta variant. Recent research has shown that infections with the delta variant put patients at a higher risk of severe disease leading to hospitalization.
Sustained genomic surveillance is important to identify newly emerging variants. Any significant clinical associations could be of great importance to attempt to contain the pandemic.
*Important notice
medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Cedro-Tanda, A. et al. (2021) "The evolutionary landscape of SARS-CoV-2 variant B.1.1.519 and its clinical impact in Mexico City". medRxiv. doi: 10.1101/2021.09.07.21262911.
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: Amino Acid, Chest Pain, Coronavirus, Coronavirus Disease COVID-19, Cyanosis, Evolution, Frequency, Genome, Genomic, immunity, Pain, Pandemic, Phylogeny, Protein, Reproduction, Research, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome

Written by
Colin Lightfoot
Colin graduated from the University of Chester with a B.Sc. in Biomedical Science in 2020. Since completing his undergraduate degree, he worked for NHS England as an Associate Practitioner, responsible for testing inpatients for COVID-19 on admission.
Source: Read Full Article